Phylogenetics
"This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison."
introduction
What is phylogenetics?
Phylogenetics is the study of the evolutionary history of a gene or an organism (1). Phylogenetics and trees are used to infer and understand relationships between related species or genes. Phylogenetics can be used to understand and reconstruct evolutionary models (1). Importantly, phylogeneomics can be used to depict and learn how a sequence has evolved (1).
How are trees built?

|
Trees are built by aligning sequences and noting similarities and differences in the nucleotide alignment (2). These sequences differences are given different scores based on the probability of the given nucleotide at a specific position in the sequence. These scores are used to calculate distances, determine branch lengths, and construct trees (2).
|
NEighbor joining method
|
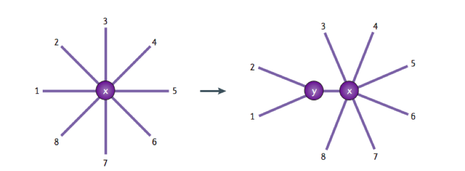
The neighbor joining method utilizes a distance matrix along with a cluster algorithm (2). The neighbor joining method begins with a star tree, where the organisms or genes in consideration are arranged in a star, depicting no evolutionary relationship (2). The most related species are then clustered together based on calculated distances (2). The process is repeated several times until a neighbor joining tree is formed (2).
Maximum Likelihood method
|
Maximum Likelihood creates a tree that focuses on probability (2). Specifically, the tree is created so that it maximizes the probability that tree is true, given the data (2). Maximum likelihood also optimizes branch lengths.
|
Average distance method
The average distance method uses the sequence alignment to determine similarity scores between related species. The algorithm then uses these scores to group the most closely related species on a branch with equal branch lengths (3). These equal branch lengths (3).
Results
SNCA sequence Alignment
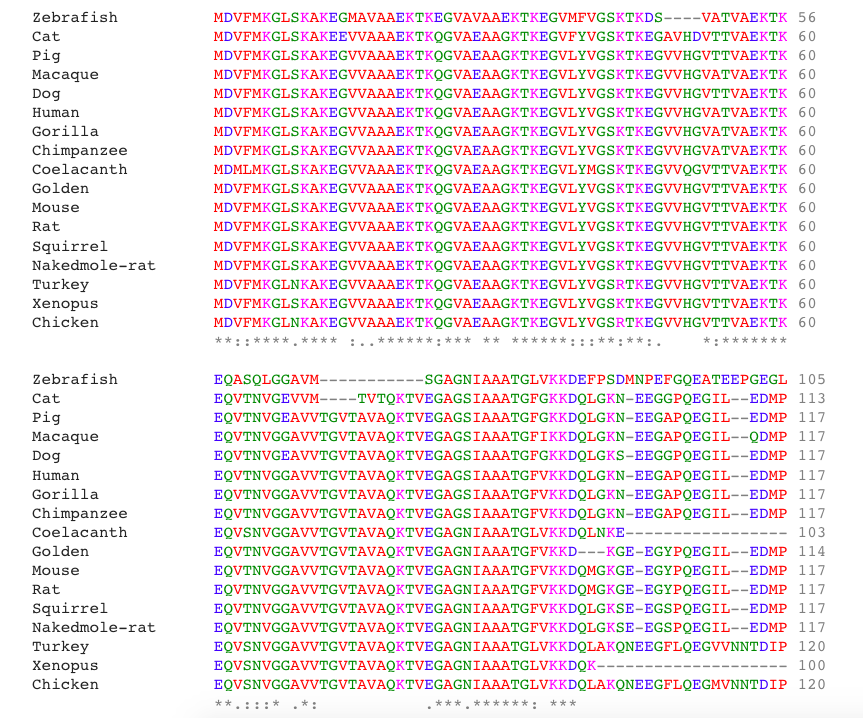
|

|
SNCA phylogenetic tree
The Neighbor joining tree
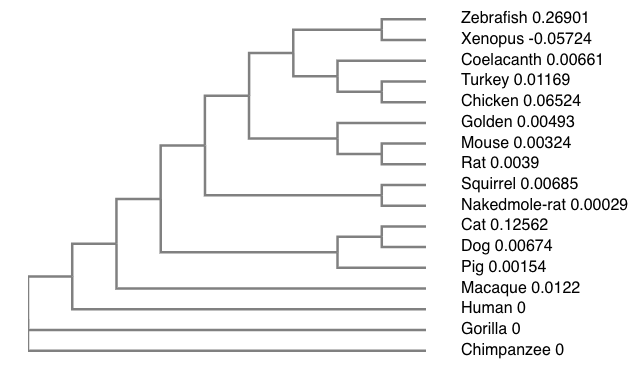
Above is a phylogenetic tree for the SNCA gene. This tree was constructed using the Neighbor joining method employed by clustal omega, and represents the evolutionary history of SNCA. Branch lengths are shown on the right, indicating nucleotide substitutions/site.
Discussion
Different tree building methods have different strengths and weaknesses. Often, several types of methods are used and the trees are then compared. This comparison allows for the most representative and most accurate tree. The neighbor joining method is often a favorite because it is fast, and is able to compare large data sets. Furthermore, based on the sequence alignment and the phylogenetic tree above, it is clear that SNCA is highly conserved among orthologs, especially in the N-terminus region of the protein. Interestingly, the N-terminal KXKEGV repeats have been found to be highly conserved among homologs and important for lipid binding (4).
References
1. What is phylogenetics?-EMBL-EBI. (2018). Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
2. Yang, Z., Ranalla, B. (May, 2012). Molecular phylogenetics: principles and practice. Nature Reviews. 13, 303-314. doi:10.1038/nrg3186.
3. Lab 2: Homology and Phylogeny-Genetics 564. Retrieved from https://genetics564.weebly.com/homology--phylogeny.html
4. 1.Siddiqui IJ, Pervaiz N, Abbasi AA. (2016). The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Scientific Reports, 6:24475. doi:10.1038/srep24475
Header photo: Retrieved from http://slideplayer.com/slide/10467135/
2. Yang, Z., Ranalla, B. (May, 2012). Molecular phylogenetics: principles and practice. Nature Reviews. 13, 303-314. doi:10.1038/nrg3186.
3. Lab 2: Homology and Phylogeny-Genetics 564. Retrieved from https://genetics564.weebly.com/homology--phylogeny.html
4. 1.Siddiqui IJ, Pervaiz N, Abbasi AA. (2016). The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Scientific Reports, 6:24475. doi:10.1038/srep24475
Header photo: Retrieved from http://slideplayer.com/slide/10467135/
